 This is
a conference given at ACA'97
in the topic SOLUTION
AND REFINEMENT FROM POWDER DATA. The full text and the slides are available
below, together with the abstract.
This is
a conference given at ACA'97
in the topic SOLUTION
AND REFINEMENT FROM POWDER DATA. The full text and the slides are available
below, together with the abstract.Standard and Special Strategies in Structure
Determination from Powder Data
Armel Le
Bail
Université du Maine, Laboratoire des Fluorures, CNRS
UPRES-A 6010, 72017 Le Mans, France.
This is
a conference given at ACA'97
in the topic SOLUTION
AND REFINEMENT FROM POWDER DATA. The full text and the slides are available
below, together with the abstract.
Abstract :
A special strategy is defined here as an alternative when the
usual way fails. In this sense, using one of the 30 or so synchrotron or
neutron facilities is a special strategy : something to try if conventional
X-rays did not succeeded or if a special accuracy was needed. Remember
that more than 15000 conventional X-ray powder diffractometers, 2000 Guinier
and 20000 Debye-Scherrer cameras may be found all around the world. The
standard powder data strategy is very similar to a small molecule structure
determination from single crystal data : one has successively to determine
the cell, the space group, to measure the structure factors, to apply Patterson
or direct methods, to complete the structure by Fourier difference syntheses
and to refine it. This requires the best of crystallography softwares and
a quite meticulous work. Lot of time can be wasted by errors in `automatic'
cell and space group false propositions. There are two obvious reasons
for failing in SDPD (Structure Determination from Powder Data), the first
is that a powder is not always made from a pure compound (one sufficient
single crystal picked in a mixture has no equivalent here) ; too low quality
data is a second reason at one step or another, regarding the problem size.
Hints have been gathered in a tutorial recently added to the SDPD-D [1]
allowing a beginner to follow the standard strategy as applied to various
cases. Applicability limits are related to diffractometer resolution, problem
size and sample quality. Their knowledge leads now to admit that a SDPD
is a routine task because failure can be easily predicted, sometimes by
just looking at the pattern. Special strategies may require more sophisticated
approaches than the above 'brute force' as the estimation of a partition
of the strictly overlapping reflections other than equal, Bayesian analysis,
rotation of known fragments inside the cell, model building, Monte Carlo
moves up to obtain optimum atom position. Works using these approaches
are currently few among the SDPD-D 300 cases. Availability in the public
domain of softwares allowing these special data treatments may increase
applications but they will logically represent a last chance when the standard
easier strategy fails. A universal and cheap crystal data bank gathering
all characterized compounds and allowing to calculate powder patterns is
desirable for avoiding structure re-determination and useless efforts.
[1] Structure Determination from Powder Diffraction - Database, A. Le Bail, available at URL http://www.cristal.org/iniref.html (1994-97).
Full text and slides :
1- Going ab initio from a powder diffraction pattern of an unknown compound to its crystal structure, this is the subject of this conference. In the ten last years the discipline has exploded in a large number of methods for data analysis so that a beginner may have difficulties in choosing the most appropriate way for solving his own problem. I will try to make a distinction between special and standard strategies. Slide 1.
2- Will be examined successively the possible radiation sources, the methods for data analysis, the reasons for failing, the limits and finally the role of databases. Percentages given will refer to nearly 300 experimental cases of which 90% were determined ab initio from powder data in the last ten years. Slide 2.
3- First of all, it is not difficult even for a beginner to identify what is standard and what is very special concerning the radiation sources. Synchrotron and Neutron radiations are very prestigious and we will see that if one expect to solve the most complex problems, the synchrotron radiation is the best choice. However it must be recalled that for many moderately complex problems, an in-laboratory Bragg-Brentano diffractometer will be quite sufficient. Indeed 70% of the published ab initio structure determination from powder data are made at home. You may have recognized the European Synchrotron Radiation Facility and the Institute Laue Langevin neutron reactor at Grenoble, France. Slide 3.
4- Powder data analysis, when the scope is to determine a structure, is made following a step by step approach. Here is a list of six operations usually realized in this order with some variants. At some steps, special strategies could be required because the standard way fails. Of course some strategies called standard nowadays were considered as quite special ten years ago. Probably most of the data analysis I call special today will be very common in ten years. We will examine briefly each step. Slide 4.
5- Each step is essential. However the preliminary step is probably the most consuming in time with 90% maybe of the whole job (in which I do not include the manuscript redaction, nor the delay for publication). What are the reasons why you should undertake a structure determination from powder diffraction data ? I am afraid I will list some evidences here. Deciding to determine a structure from powder data supposes that you have synthesized a compound, that you failed in a positive identification from databases. Sometimes you may not identify the compound before doing an indexation of the powder pattern. Even at this stage you may prefer to try to make larger crystals by modifying your way of synthesis. A structure determination from single crystal data is always preferable to a powder work. Nevertheless a structure determination from powder data by standard strategies may be quite fast so that the question « should I waste so much time for obtaining a more accurate result » may have a negative response. Most people think that this is a bit terrific for structure data quality because powder diffraction leads necessarily to mediumly accurate results. This list of chapters is taken from the beginning of a tutorial on structure determination from powder data available on Internet. Slide 5.
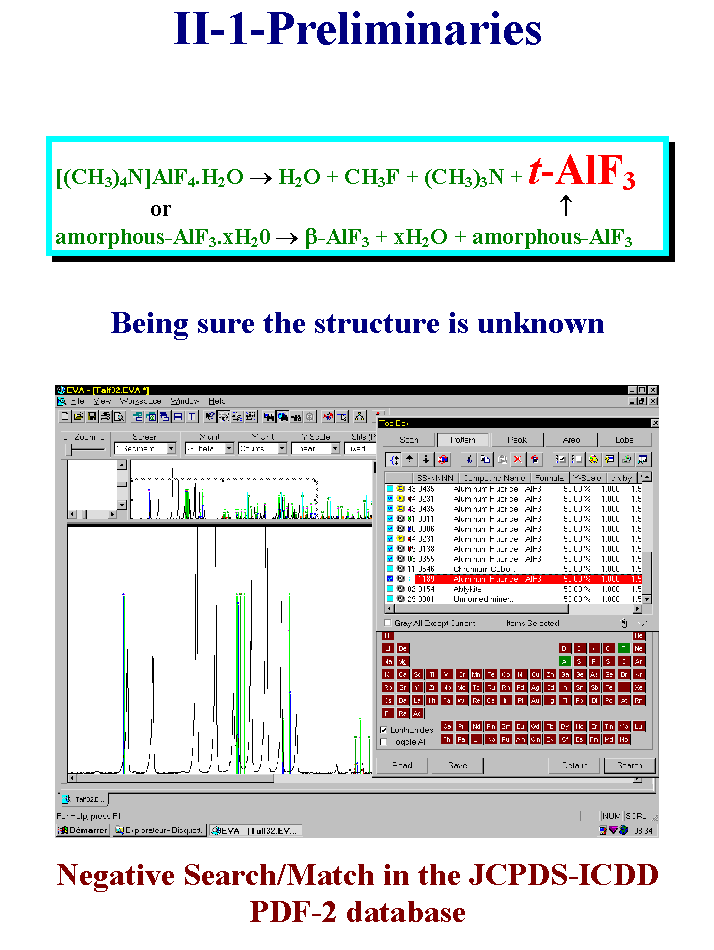
6- OK, just an example of what I mean by « preliminaries ». An unknown new compound came either from the dehydration of an amorphous hydrated aluminum fluoride or from the thermal treatment of a tetra-methyl-ammonium aluminum fluoride salt. I show here the negative search-match result from the PDF-2 database by using a commercial software. You can imagine the work behind this disclosing of a new AlF3 variety. Its composition was established because it transformed into the high temperature cubic form on heating without any new mass loss. Please note that the t-AlF3 powder pattern is still not in the 1996 version of the PDF-2 database in spite of the structure published in 92 in the Journal of Solid State Chemistry. You can find it now exclusively in the Inorganic Crystal Structure Databank (ICSD). Slide 6.
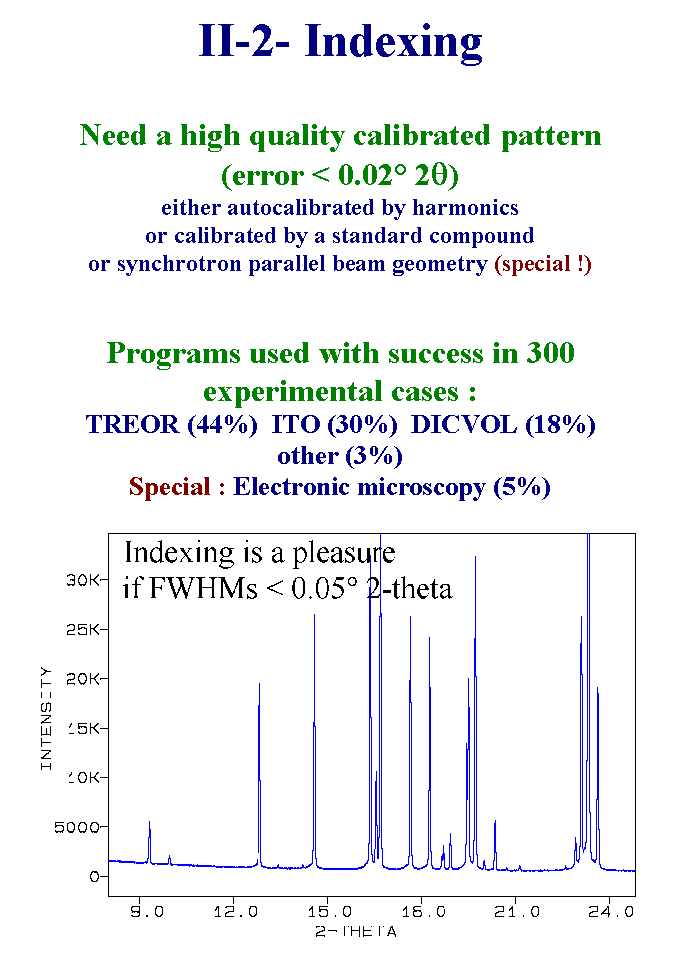
7- Up to now, there is nothing special, just standard job. Doing the preliminary job directly by recording synchrotron or neutron data would not make sense. Being sure that the compound is unknown may need that the indexation is done in order to verify in all databanks and by all bibliographic means that you are really not determining a structure already known. The chemical composition should suffice for that, however two precautions are better than one because the chemical composition estimation can be in error sometimes. So that indexing may be considered as being included in the preliminary work. Well, what are the recommended indexing procedures those days ? Among the 300 or so experimental cases, three sofwares were used with considerable success which are TREOR, ITO and DICVOL. Testing your data with these three softwares is the best thing to do before to go to the next step. Indexing can be a real pleasure if your data have Full Width at Half Maximum less than 0.05° 2-theta. The pattern shown here corresponds to the famous cimetidine powder pattern as measured from synchrotron data. It is possible to do as well from in-Laboratory instruments equipped with variable slits. Trying to index from bad data is waste of time. Slide 7.
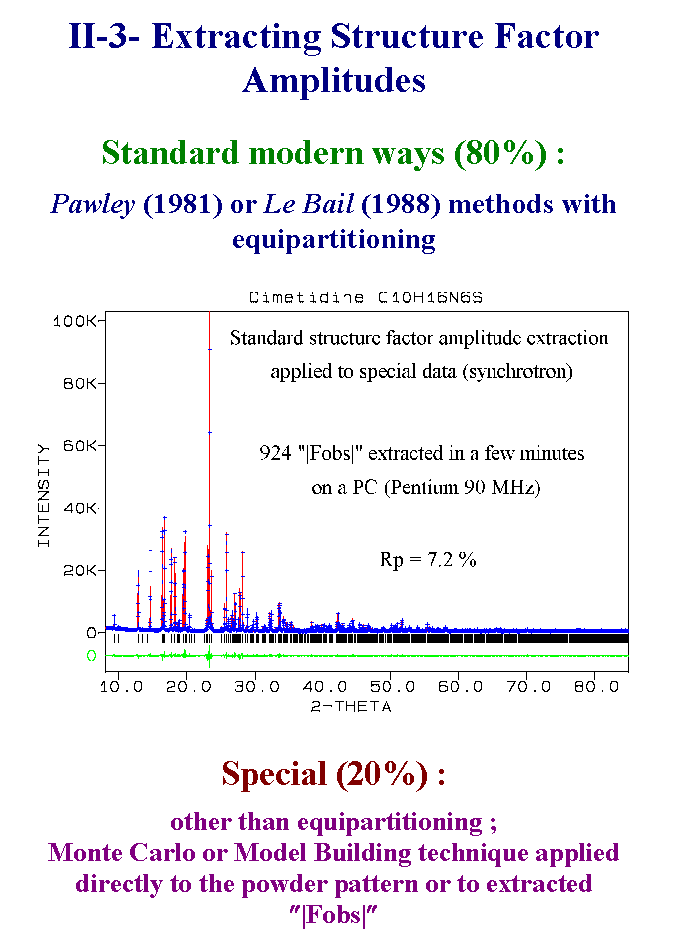
8- The standard strategy is then to extract the structure factor amplitudes by the Pawley or Le Bail methods. Thanks to these cell constrained methods, one thousand of structure factors can be obtained in a few minutes. Of course most of them will be quite false. The strictly overlapping reflections are generally equipartitioned. Some special methods may be applied here. For instance the equipartitioned reflections may be modified according to suggestions obtained from the non-overlapping reflections. On another hand, some special methods may avoid this extracting structure factors stage by a direct comparison of the observed pattern with the pattern calculated from some model building process. The same model building methods can be applied also to the extracted structure factors. Slide 8.
9- Solving the structure means obtaining an initial model sufficient for starting a refinement allowing to complete the structure by Fourier syntheses. Direct and Patterson methods were used in 90% of the 300 experimental powder cases. They were applied on the full dataset or on more or less reduced datasets as a function of the overlapping degree, sometimes exclusively on the unambiguously indexed reflections. Only 10% are concerned by other approaches. It may be considered as normal that the high resolution synchrotron data could allow the simultaneous location of a high number of independent atoms by applying the direct methods. For instance the cimetidine whole dataset allows to locate all the 17 non hydrogen atoms from 900 reflections. In the first study, the SIR program was claimed to be the only program giving this result. I have a different conclusion because at least SHELXS works as well. More surprising is the fact that problems as complex as the cimetidine can be also solved from conventional X-rays or even neutron data with full widths at half maximum up to seven time larger. Slide 9.
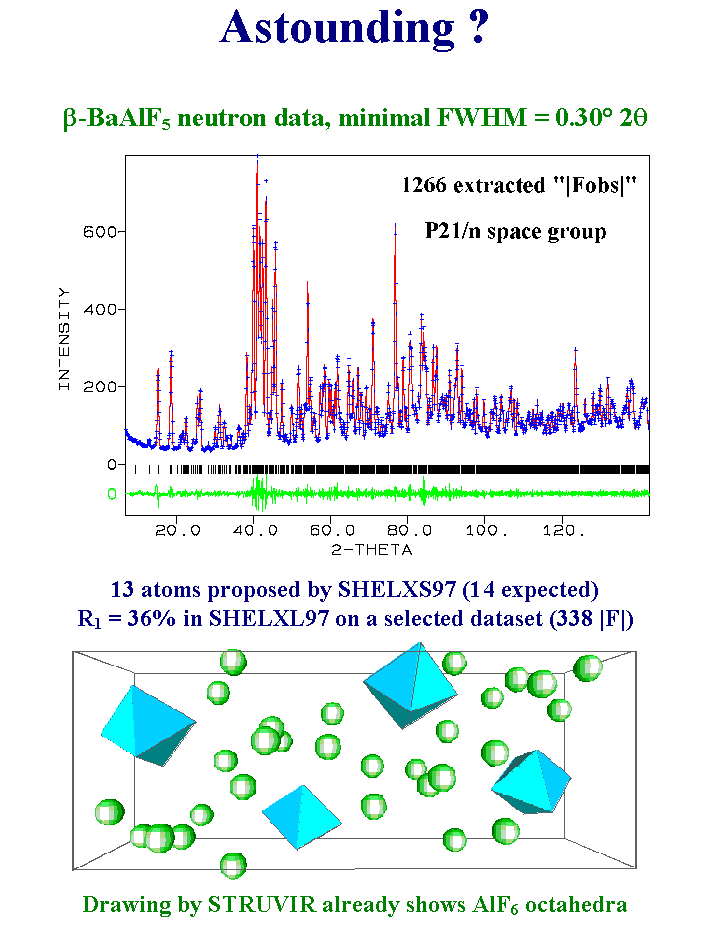
10- If we look more closely at the worst of the above cases, the barium aluminum fluoride neutron data, we cannot seriously believe that more than 100 or 200 reflections make really sense among the 1266 extracted ones. Nevertheless it is a fact that the direct methods applied to the whole dataset give 13 oF the 14 expected atom sites. Of course, for testing the model, one should make a refinement on a dataset reduced by the elimination of the reflections overlapping too much. Taking 300 of the best reflections, one obtains a reliability factor not very satisfying, however the structure drawing by a software recognizing polyhedra shows undoubtedly that a considerable starting model has been disclosed. How this astounding result is possible ? Well, my theory is that the false reflections are randomly false when obtained by the Pawley or Le Bail methods with equipartitioning so that their contribution is mainly to increase a flat background on which only those reflections really associated with an ordered model produce peaks located by the direct methods. We will see later the limits above which there would be no hope to solve a structure. Of course, this example was not in reality solved from the neutron data. I like difficulties but not to this level. The two barium atoms were located by either the Patterson or direct methods from X-ray conventional data. The neutron data were used for a final improvement of the atomic coordinates accuracy. Slide 10.
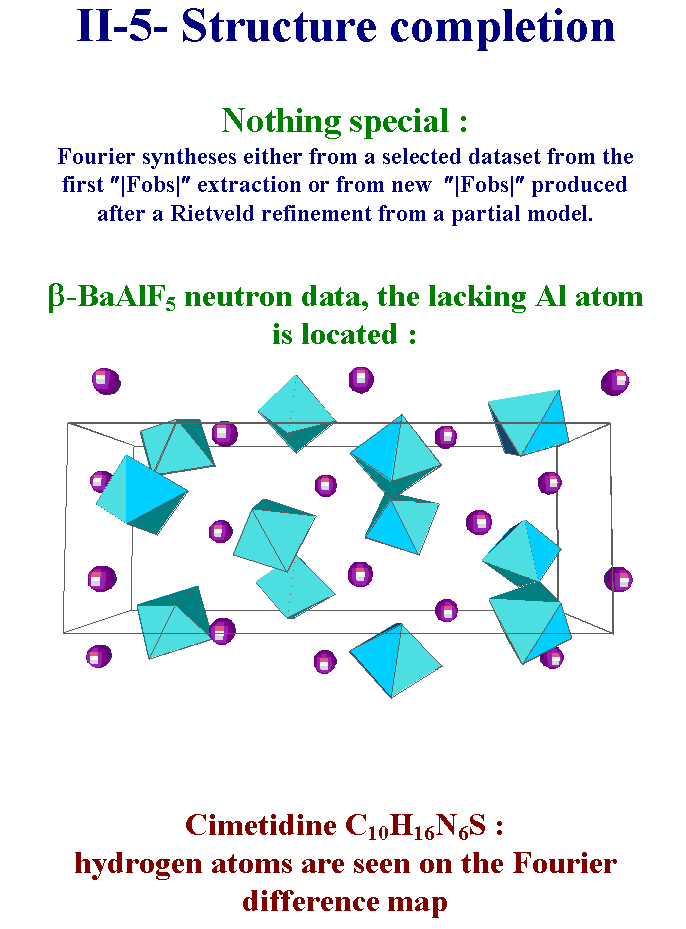
11- When the whole structure has not been revealed by direct or Patterson methods one has very classically to complete the model by Fourier syntheses applied on either a reduced dataset from the first structure factors extraction or from a new extraction at the end of a Rietveld refinement. In the case of the BaAlF5 structure, the lacking aluminum atom is easily located. For the cimetidine, hydrogen atoms appear on the Fourier difference map. These Fourier syntheses are realized by using standard softwares for refining single crystal data like SHELXL. Slide 11.
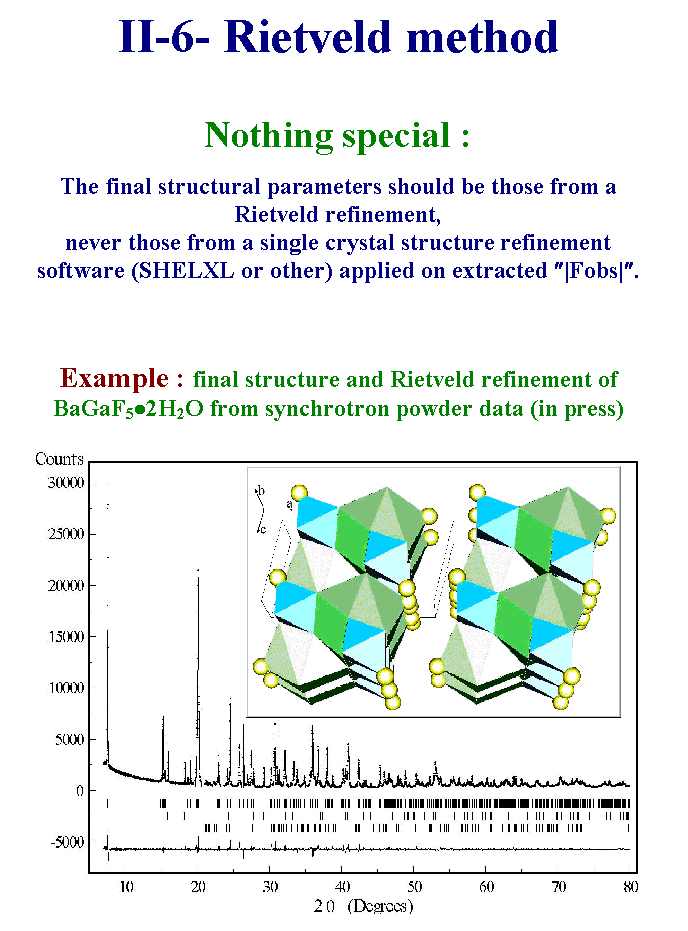
12- The Rietveld method should always be the last step giving the atomic coordinates for publication. Here is an example of the final Rietveld refinement on a new barium-gallium fluoride di-hydrate as determined from synchrotron powder data. So, this example makes use of standard methods applied to special synchrotron data. The philosophy is to use special strategies when standard one failed. In this case the problem was the difficulty to make a pure compound so that the synchrotron high resolution was needed because of the increase of complexity due to the presence of two unavoidable impurities. We will examine the two main reasons for failing in a structure determination from powder diffraction data. Slide 12.
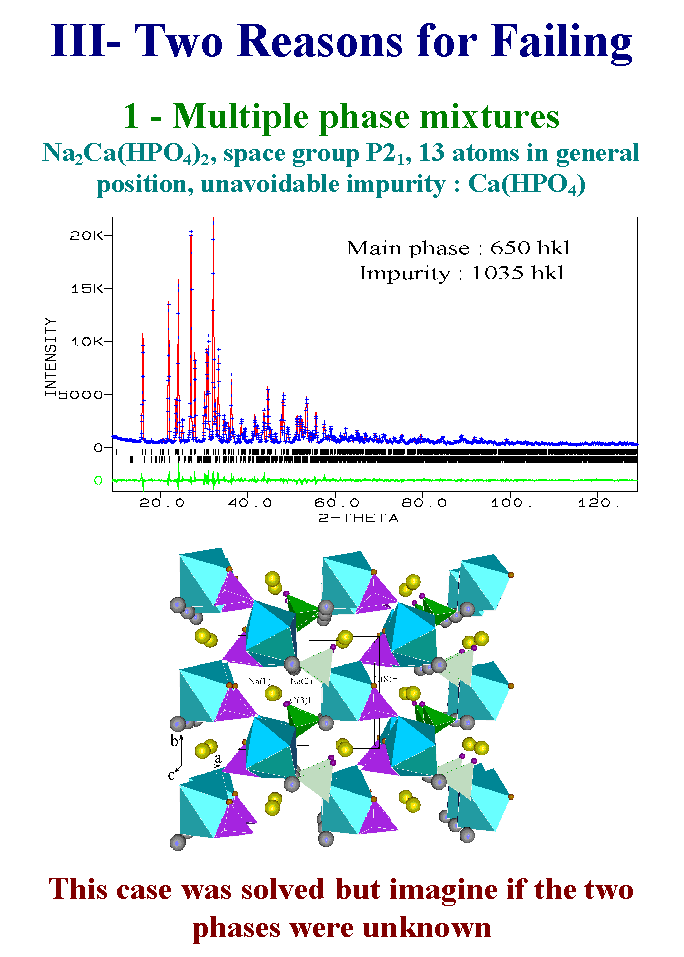
13- The first reason is associated with the difficulties in preparing a pure compound. This does not matter when single crystals are available because just one crystal picked in a mixture could be sufficient. Here is another example of an unkwown whose structure was solved in spite of the presence of one unavoidable impurity. Fortunately, the impurity could be identified and its structure was known so that its contribution is calculated and does not add new refined parameters but a scale factor. You can imagine if you had a mixture of two or three unknown compounds simultaneously present in your preparation. Without other data like the cell parameters obtained from electronic microdiffraction you will fail. Slide 13.
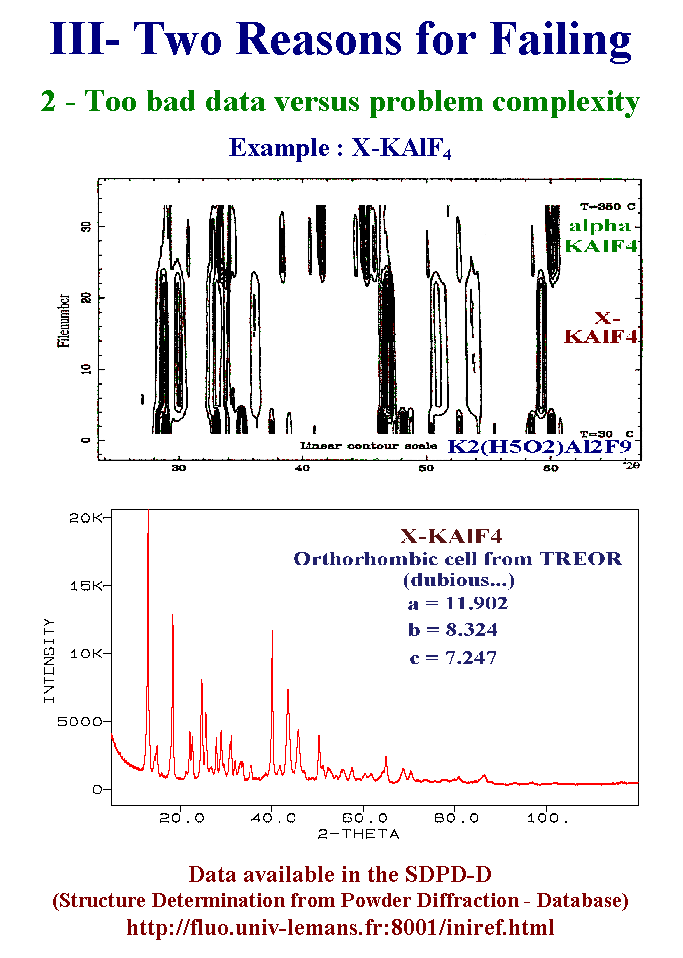
14- The second reason for failing is that your data are too bad versus the problem complexity. Here is an example which has been put in the public domain so that everyone wishing to try to solve it, is welcome and will be the unique author of the paper in case of success. This new compound is made by the dehydration of a potassium aluminum fluoride. Its formulation is simple and we are sure of it because it transforms at 220°C into a known form of potassium aluminum tetrafluorine without any further mass loss. The new compound is microcrystalline so that the reflections full width at half maximum are four time larger than those of the initial hydrated compound or those of the high temperature final form. In such a case, using synchrotron data is of no help. Moreover, the sample does not resist to an electron beam. So that indexing is absolutely not sure. A proposition was made which will remain dubious until a successfull structure determination. Up to now, I noticed 30 persons who downloaded the data from my Internet Web server since three months, nobody succeeded in a structure proposition either from standard or special strategies. Slide 14.
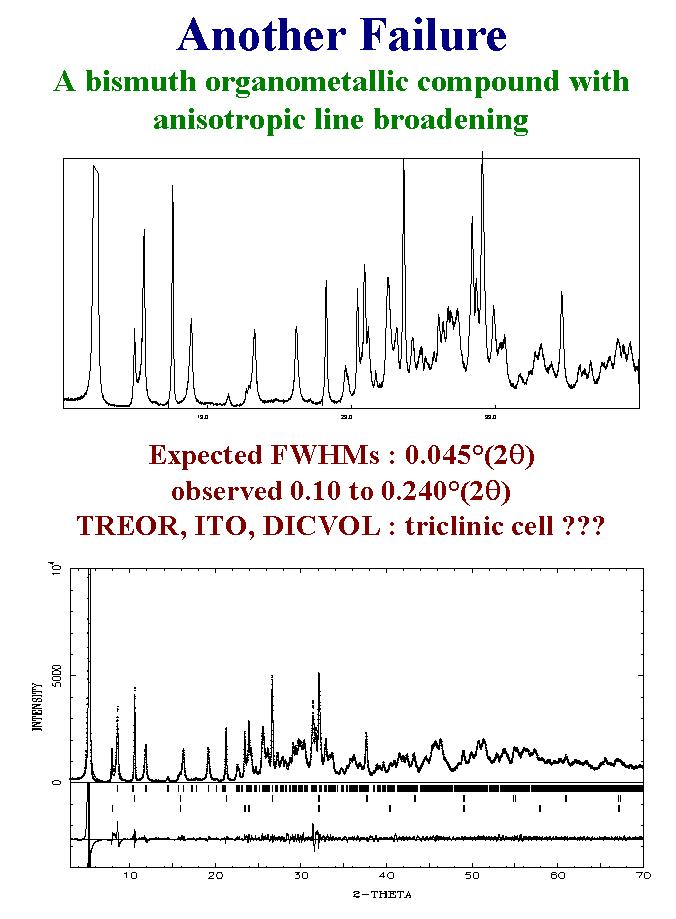
15- Another example showing the limits of the standard strategies as well as those of special strategies is that of a bismuth organometallic compound. The conventional X-ray powder pattern is made with a diffractometer equipped with variable slits so that full width at half maximum as low as 0.04 two theta degrees could be expected. In fact there is X-ray line broadening and moreover anisotropic so that the observed full width at half maximum are ranging between 0.1 and 0.24 two-theta degrees. Just looking at such a pattern should discourage anybody to try to do the job. Of course the indexation propositions were very dubious. Attempting to extract structure factors in the hypothesis of the cell propositions never lead to anything up to now. This problem will have a solution when the sample crystallinity will be improved. Unfortunately, many attempts for making better crystalline samples failed. Slide 15.
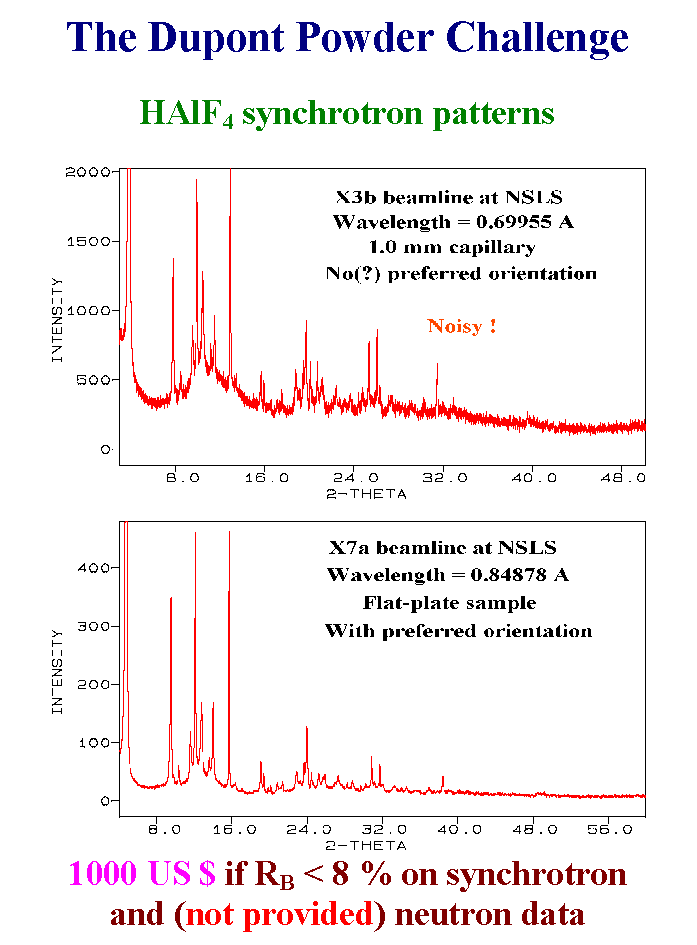
16- May be you have seen recently at the crystallography Newsgroup on Internet the announce of « The Dupont Powder Challenge ». The Dupont specialists failed to determined the HAlF4 structure from synchrotron and neutron powder data. So that they offered 1000 US$ to the individual or team which could attain a Bragg reliability factor lower than 8% on these synchrotron data with a model that would also give 8% on the neutron data. However they did not give the neutron data. Was it really a challenge and were special methods requested ? Of course I was interested in the challenge more for glory than for the small award. As you see there is one very noisy pattern, the other being affected by some preferred orientation. It took one day for being convinced that I had an acceptable cell and space group propositions. Then another day for extracting structure factors and trying to determine the structure by direct methods. A complete proposition without the hydrogen atoms came and three days more were needed for attaining the 8% requested reliability. There are many problems associated with anisotropic line broadening, impossibility to find exactly the hydrogen atom position from X-ray data due to disorder on HF molecules, very high background. So that the challenge is still on the road and the Dupont�s people decided to send the neutron data 3 weeks after the challengers will propose a model from the X-ray data. Slide 16.
17- OK, the proposition I obtained has certainly a large part which is correct because it cannot be a haphazard to have aluminum atoms in octahedral coordination forming layers built entirely from the direct methods. You will have exactly two seconds for seeing the model because if one thousand dollars is not enough it is better than nothing. Here is the model : one, two, sorry.
The slide cannot be shown here...
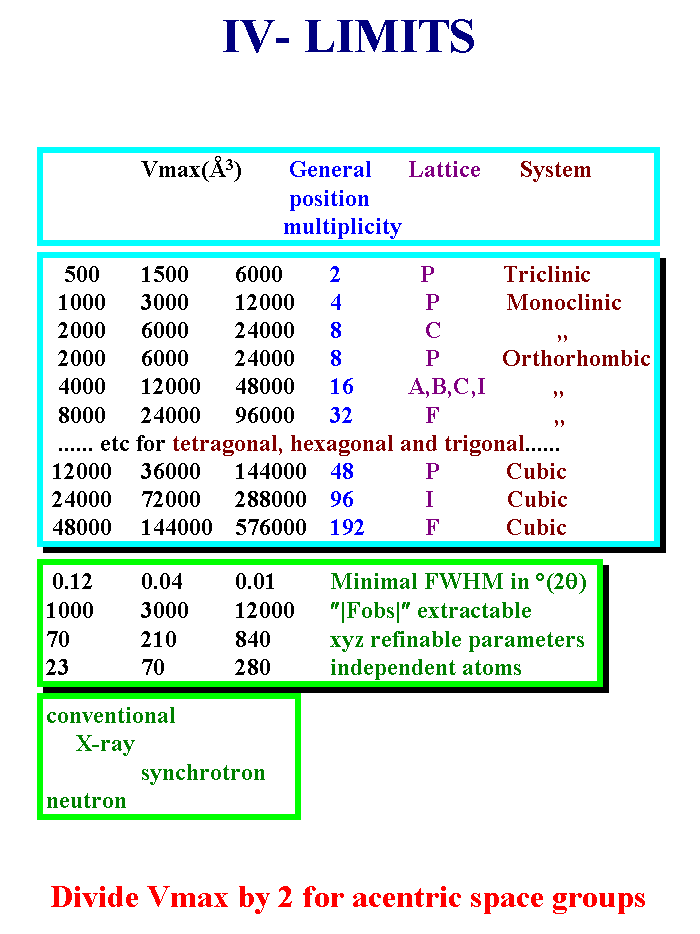
18 - So, the Dupont powder challenge finally does not give the limit of what is possible from powder data. One can estimate the limits by looking at the experimental cases already solved as a function of the data resolution in full width at half maximum. Depending on the symmetry one may expect to determine structures corresponding to cell volumes as large as 6000 Angstroem-cube for triclinic to half a million Angtroem-cube for a F-centered cubic compound by using synchrotron data with the highest resolution, that is to say, 0.01 2-theta degrees as full width at half maximum, 12000 extracted reflections. Count on twelve time less with neutron or conventional X-ray data or medium values for the best of conventional x-ray data or the worst of the synchrotron sources. Of course these values suppose that your sample is well crystallized, with no special line broadening. I hope you find these limits impressive. To my knowledge, cases with 300 independent atoms have not yet been determined and refined. The current maximum value is near of seventy independent atoms. Slide 18.
19- Now from my own experience I believe that an exhaustive database containing powder diffraction patterns is urgently needed. Such a database, exhaustive, is the key for avoiding wasting time and resources. I have personnally wasted a lot of time, maybe one year of my life because of unsuccessfull research at the preliminary identification stage leading eventually to useless structure redetermination. Due to a monopolistic position the International Center for Diffraction Data concentrates all my hopes. It seems that unprecedented efforts are currently made for including some lacking powder patterns as calculated from crystal data actually listed in the other well known databases. That is really good news ! May be in the future, it will be less frequent to read in manuscripts dealing with structure determination statements like : « once this work was completed we were informed that a similar job was done by another research team ». This problem has also attained structures determined from powder data and you can find among the 300 experimental cases some which were determined twice from powder data (t-AlF3 for instance with 3 years between the two publications) or you can find isostructural compounds for which in principle an ab initio determination was not really required. Slide 19.
20- As a conclusion, a question generally asked after a conference on this subject is "what is the minimal size of the single crystal below which you prefer to determine a structure from powder data" ? Well, my opinion is that very few good structure determinations from very small single crystals with moderatly complex structures have been really solved either from electronic microscopy or synchrotron data. I hope I have convinced you that powder diffraction is really competitive and represents a very good alternative. Powder diffraction requests some expertise and I recommend first to determine some structures from single crystal data before to try from powder data. You have understood that in my opinion, standard strategies in structure determination from powder data are similar to the common strategies developed for solving moderately complex structures from single crystal data. An advantage of the methods I have described is that the tools are in the Public Domain. There is no doubt that special strategies are requested if the standard ones failed. Many of these special strategies are emerging that could be renamed standard if their use increase, and if tools are made simple and available. Slide 20.
21- More information may be found in the Structure Determination from Powder Diffraction - Database giving references comments and statistics for 300 experimental cases. A tutorial is included explaining how to realize an ab initio structure determination by standard methods. Five examples are fully explained from the beginning to the end with all the intermediate results available. Data are from conventional X-ray diffraction as well as from synchrotron X-ray and neutron diffraction. The Internet address is given here and in the conference abstract. Slide 21.
For more information on special strategies, see the other conferences on this topic.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}