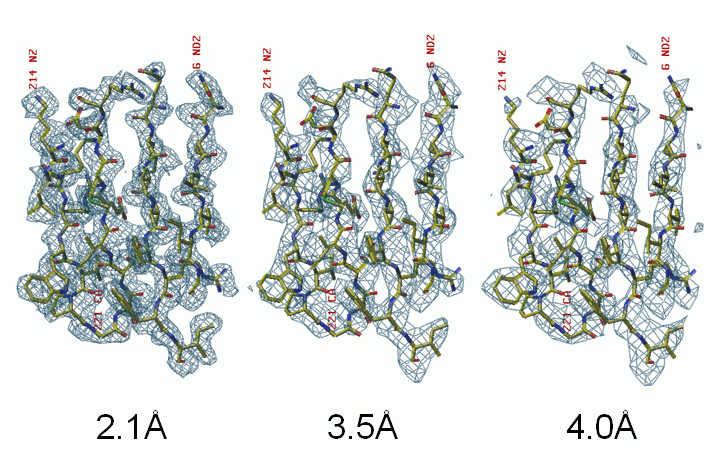
This study is to probe the low resolution limit that allows a successful direct-method SAD phasing. Sulfur-SAD data of the 69 kDa protein TT0570 were collected with in-house Cu-Kα radiation at 1.8Å resolution. Its truncated subsets at 2.1, 3.5 and 4.0Å resolution were used in the test. The sulfur substructure was solved at each resolution independently. The data has an expected Bijvoet ratio <|ΔF|>/<|F|> ~ 0.56%. In the 2.1Å case, a single run of OASIS+DM+ARP/wARP led automatically to a model containing 1178 of the total 1206 residues all docked into the sequence. In the 3.5Å case, two cycles iteration of OASIS+DM+RESOLVE (build only) resulted in a model with more than 700 residues, 58 among them were docked into the sequence. The corresponding electron density map is traceable by an experienced worker. In the 4.0Å case, a single run of OASIS+DM+RESOLVE (build only) gave a somewhat degraded map, which failed to be improved by further iteration but still provides useful information.
By D.Q. Yao1, 2, 3, Y.X. Gu1, C.D. Zheng1, Z.J. Lin4, H.F. Fan1 & N. Watanabe5
1Institue of Physics, Chinese Academy of Sciences, Beijing 100080, China
2Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
3National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei
230026, China
4Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China
5Faculty of Advanced Life Sciences, Hokkaido University, Sapporo 0600810, Japan