The program Bond_Str and its GUI GBond_Str
(Version March 2005, J. Rodríguez-Carvajal - LLB)
The program Bond_Str calculates distances and angles in
crystal structures. It is based in old Fortran 77 versions but is has been
completely re-written and it is now based in CrysFML (Crystallographic Fortran
95 Modules Library). CrysFML contains in its "Atom_Module" (in file CFML_Atom_Mod.f90), procedures making all
calculations. The main program calls the two procedures "Calc_Dist_Angle_Sigma" and "Calc_BVS" to perform the calculations.
The most reliable bond-valence parameters
(based in the file bvparm.cif from I.D. Brown) are stored in CFML_BVpar.f90, but, alternatively, user-given
bond-valence parameters can be read form the input file instead of using the
internal parameters.
The program needs an input file that can be
either a standard CIF file or a CFL file containing just the necessary
structural information and the ionic species. A CFL file is a file with a
format that is recognized by all programs based in CrysFML. They have the
extension *.cfl and are in
free format. The different items are recognized thanks to keywords. A CFL file
can be generated from a CIF file just running Bond_Str.
Alternatively, the GUI GBond_Str program can be used directly to
convert CIF files to CFL files. Remember that information about the chemical
species (ionic oxidation states) is not always included in CIF files, so the
user has to include it in the appropriate place in the atom string (see below)
if he(she) wants to make bond-valence calculations.
An example of CFL file is given below.
The program can be invoked from the command
line together with the code (name without extension) of a CIF or CFL file. The
program looks first for the existence of a CFL file with the given code, if
there is no CFL file it looks for a CIF file.
All it is needed to know about the input files
and running the program is explained in the following two examples.
Example 1:
A CIF file, called myfile.cif, exists in the current directory
(and there is no file called myfile.cfl); the program can be invoked as follows:
Current_directory>
Bond_Str myfile <cr>
<cr> corresponds to carriage return
(ENTER key)
The screen output is the following:
==============================
====== PROGRAM BOND_STR ======
==============================
***********************************************************************
* Distances,
angles and Bond-Valence Sums from *.cfl
or *.cif files *
***********************************************************************
(JRC
- LLB, version: March 2005 )
=> A CFL-file has been generated from CIF ->
CFL_file.cfl
This file may
be used to add instructions for BOND_STR
=> Bond-Valence calculations impossible: ionic
charges must be provided!
=> Normal End of: PROGRAM BOND_STR
=> Results in File: myfile.bvs
In the input CIF file there is no information
to calculate the oxidation state of the different ions, so bond-valence calculations
are not performed.
Only distances up to 3.2 angstroms are
calculated for the current structure. A file called CFL_file.cfl has also been generated and the
user can include the necessary information to perform the complete calculations
in further runs (using the CFL file instead of the CIF file). This can be done
more easily using the GUI GBond_Str and importing a CIF file that is automatically transformed into a CFL
file.
Example 2:
A CFL file, called mfepo5.cfl, exists in the current directory;
the program is invoked as follows:
Current_directory>
Bond_Str mfepo5 <cr>
. . . .
The screen output is the following:
==============================
====== PROGRAM BOND_STR
======
==============================
***********************************************************************
* Distances, angles and Bond-Valence Sums from *.cfl or *.cif files *
***********************************************************************
(JRC - LLB, version: March 2005 )
Subroutine Calc_BVS (JRC-LLB, version: March-2005)
Title: Summary of Bond-Valence calculations for file: mfepo5.cfl
Atom Coord D_aver Sigm
Distort(x10-4)
Ni 6.00 2.0801(
6) 3.127 2.000 1.906(
3)
Fe 6.00 2.0470(
21) 72.020 3.000 3.015( 20)
P 4.00 1.5301(
20) 0.967 5.000 5.064( 28)
O1 4.00 2.0251(
31) 15.651 -2.000 1.784( 19)
O2 4.00 2.0538(
17) 231.644 -2.000 1.929( 18)
O3 2.00 1.7029(
32) 132.640 -2.000 2.032( 21)
O4 3.00 1.8603(
7) 156.068 -2.000 2.120(
5)
=> Old Global Instability Index
(
GII=SQRT{SUM{|BVS-abs(q)|^2}/Num_Atoms} ) = 10.71 /100
=>
Normalized GII(a)= SUM {|BVS-abs(q)| *mult}
/N_Atoms_UCell = 9.15 /100
=>
Normalized GII(b)= SUM {|BVS-abs(q)| *mult/abs(q)}/N_Atoms_UCell = 4.31 %
=>
Normalized GII(c)= SQRT{ SUM
{|BVS-abs(q)|^2*mult}
/N_Atoms_UCell}= 10.88 /100
=> Normal End of: PROGRAM BOND_STR
=> Results in File: mfepo5.bvs
The screen output is the content of the summary
file mfepo5_sum.bvs.
All details are in the output file mfepo5.bvs
The content of the CFL corresponding to the
above calculation is the following:
------------------- Start of the mfepo5.cfl file ---------------------
Title NiFePO5
!
a b c alpha
beta gamma
Cell
7.1882(2) 6.3924(2) 7.4847(3)
90.000 90.000 90.000
!
! Space
Group
Spgr P n m a
! label Spc x y z Biso occ
Atom Ni NI2+
0.0000 0.0000 0.0000 0.74
0.5
Atom Fe FE+3
0.1443(9) 0.2500 0.7074(2) 0.63
0.5
Atom P P5+
0.3718(9) 0.2500 0.1424(2) 0.79
0.5
Atom O1 O2-
0.3988(9) 0.2500 0.64585(2) 0.71
0.5
Atom O2 O-2
0.19415(4) 0.2500 0.0253(4) 0.70
0.5
Atom O3 O-2
0.0437(2) 0.2500 0.4728(2) 0.83
0.5
Atom O4 O-2
0.3678(2) 0.0566(1) 0.2633(2)
0.77 1.0
! Instructions for Bond_STR
DISTANCE !
Calculation and output of distances and angles
!RESTRAINS !
Uncomment for restraints file for FullProf
DMAX 3.4
2.7 ! Fixing maximum distances dmax_dis
and dmax_angl
!
For angle calculations dmax_angl /= 0 (defaults: 3.2 0.0)
-------------------
End of the mfepo5.cfl file
---------------------
Notice the way of giving the oxidation state of
the ions: the name of the element followed by +/-n or n+/- being "n" the assumed valence. Notice
also that the standard deviations can be given in parenthesis (as usual) but
immediately following the last number. No space is permitted between the value
and its standard deviation. The minimal set of keywords in a CFL file for being
used as input of the program Bond_Str are: cell, spgr and atom. They are case insensitive.
The symbol "!" is used as a comment.
The items following an atom keyword are: Label of the atom, element or species,
fractional coordinates x, y, z, isotropic displacement parameter (Biso) and occupation factor
(proportional to the multiplicity of the site, e.g. occ=m/M).
Two more items can be given: magnetic moment
value and ionic charge as real values. If instead of the ionic species only the
element symbol is provided the two additional items are needed: even if the
magnetic moment is not used it should be given. An alternative atom-line
corresponding to the first line in the above example can be written as follows:
! Label
Element x y
z Biso occ
MagM Charge
Atom ni ni
0.0000 0.0000 0.0000
0.74 0.5 1.80
2.00
The
commented keywords DISTANCE and RESTRAINS in the above examples do not need numerical values. They just instruct
the program to change the output with respect to the default values.
The keyword
DMAX is for
limiting the distance and angle calculations.
Two real
values (dmax_dis and dmax_angl) are needed for DMAX.
Be careful
not giving a high number for dmax_angle because the number of possible angles between three atoms that are at
or below a distance dmax_angl increases strongly with dmax_angl.
If
user-given bond-valence parameters are to be provided, the instruction in the
CFL file is as follows (for instance in the case of La3+-O2-
and Mn3+-O2-):
BVPARM LA+3 O-2
2.172 0.370
BVPARM MN+3 O-2
1.760 0.370
Notes
about the GUI GBond_Str
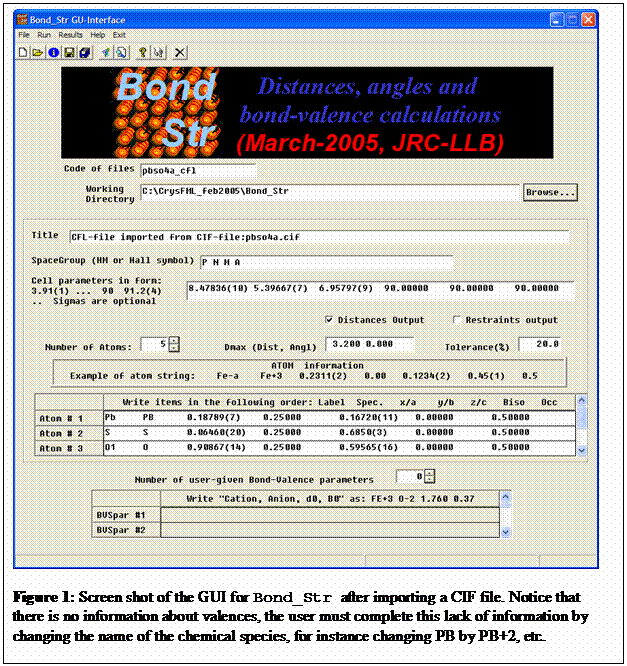
The program GBond_Str is a GUI for running Bond_Str without direct editing the input
file. The interface has only a single window, except when the internal editor
is invoked to visualise the results. The aspect of the interface after importing
a CIF file is shown in figure 1. It may be used to transform CIF files to CFL
files with just a click as shown in figure 2.
Notice that the user can provide bond-valence
parameters by filling the appropriate box that is activated as soon as one
increases the number of user-given bond-valence parameter that is initially put
to zero.
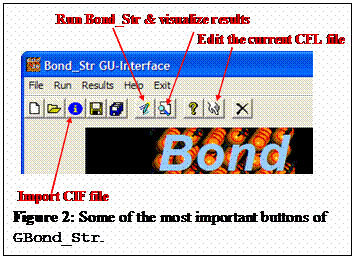 The program GBond_Str is just a tool for manipulating CIF and CFL files. There is no
calculation inside GBond_Str. When the user clicks on the run button (or select Run in the menu) the
program saves the current CFL file and invokes Bond_Str with the code of the current file
as argument. The normal output of Bond_Str that is directed to the screen when run in a
DOS-like window shell is not seen. As soon as Bond_Str finishs the calculations GBond_Str takes the control and edit the
output file from Bond_Str.
The program GBond_Str is just a tool for manipulating CIF and CFL files. There is no
calculation inside GBond_Str. When the user clicks on the run button (or select Run in the menu) the
program saves the current CFL file and invokes Bond_Str with the code of the current file
as argument. The normal output of Bond_Str that is directed to the screen when run in a
DOS-like window shell is not seen. As soon as Bond_Str finishs the calculations GBond_Str takes the control and edit the
output file from Bond_Str.