Results for Sample 2
Searching for an unknown organic compound, the tetracycline hydrochloride was first believed to be adequate from a recent publication (F.N. Blanchard, Powder Diffraction, 4 (1989) 103-105). This work states that the previous data were limited to an unindexed powder pattern (JCPDS-ICDD 13-898). The new work indexed the pattern and suggested two possible space groups (P212121 and P21212). Searching for structural data in the CSD databank produced a link to the Powder Diffraction reference and also shown that a crystal structure was determined (K. Kamiya, M. Asai, Y. Wada and M. Nishikawa, Experientia, 27 (1971) 363) under the name achromycin hydrochloride. Nevertheless, no 3D atomic coordinates were available (which is amazing). Thus it was concluded, that tetracycline hydrochloride finally represented a good sample for test, after all, given that so many already known structures served previously for testing methods. A search match procedure with the PDF-2 ICDD-JCPDS database would be positive (39-1987) and reveal the name of the compound. The molecular formula would be then very easy to find from a great variety of sources. For instance in the online Aldrich catalog. Typing "tetracycline" as keyword with the Yahoo search engine revealed 10613 links ! One can even found a .mol file and a .pdb one !
We thus tried to solve it, since the atomic coordinates were unavailable,
first looking if an isotypic compound could not be located. The CSD databank
presented two possible data sets with similar cell parameters and same
space group (P212121). These are the beta-6-deoxy-oxytetracycline
hydrochloride C22H24N2O8HCl
(DXXTEC) with a = 11.638 A, b = 12.177 A and c = 15.840 A (J. Bordner,
Acta Crystallogr. B35 (1979) 219) and the oxytetracycline hydrochloride
C22H24N2O9HCl (TERMYC) with
a = 11.19 A, b = 12.49 A and c = 15.68 A (H. Cid-Dresdner, Z. Kristallogr.
121 (1965) 170). Let us recall the tetracycline hydrochoride cell parameters
here : a = 10.983 A, b = 12.861A and c = 15.728 A. Both above data
were used as starting coordinates for a Rietveld refinement on the aldxconv
data (conventional X-rays). Only the non-hydrogen atoms were kept in the
2 starting models, and some sligth modifications were done for being consistent
with the molecular formula (the O5 atom in TERMYC was suppressed and the
same O5 atom in DXXTEC was moved to the O6 position). Trying to refine
without constraint did not led to any satisfying result. Using soft constraints
on interatomic distances, the refinement from the DXXTEC data blocked to
high reliability values, whereas the TERMYC data started at RB
= 35% after scale factor refinement, and rapidly converged
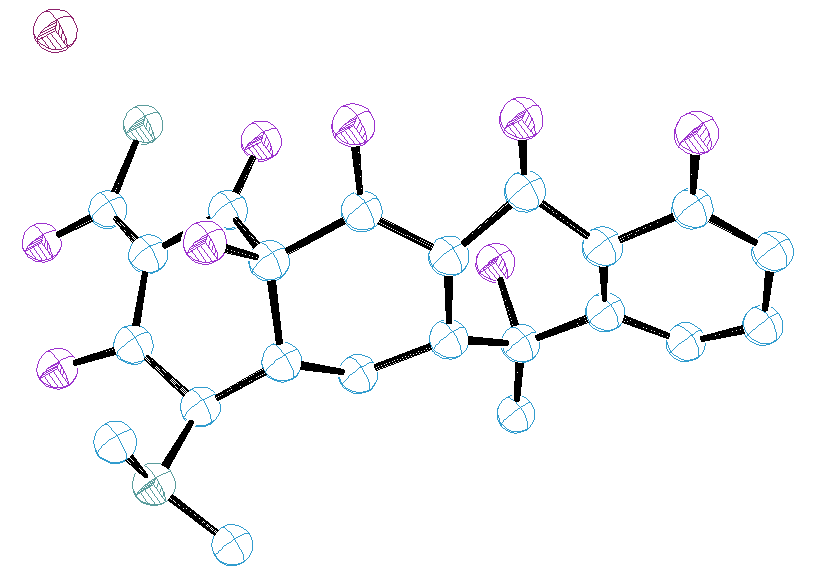
to Rp = 9.32, Rwp = 12.5 (background subtracted), RB = 4.6 %
(see the plot by DMPLOT and the Ortep
structure drawing). Then, the constraints were removed
and the reliabilities dropped to Rp = 8.73, Rwp = 11.7 and RB
= 3.7. The problem was that interatomic distances tended to be out of usual
values : it was concluded that the resolution was not sufficient.
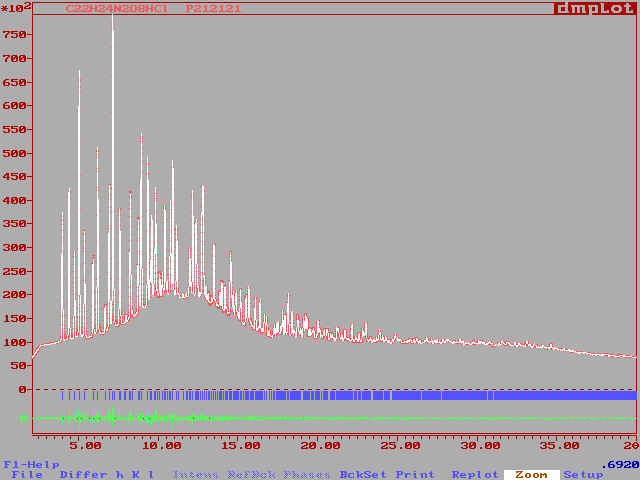
Then the synchrotron pattern was recorded, expecting a better resolution
(which is not really the case due to the short wavelength retained (0.692
A) by the operator who did not wanted to select a higher wavelength for
reasons of possible harmonic contamination). Here are the test results
for the application of conventional direct methods (i.e. from raw extracted
factors, possibly on reduced datasets). The background was estimated manually
from the aldsync pattern with the DMPLOT program. The program FULLPROF
was then used (Le Bail method). Links are given to files renamed as .txt
files :
{kind=link}
{kind=link}
- aldsync.pcr : FULLPROF instruction file
- aldsync.sum : output summary
- aldsync.fou : final extracted "|Fobs|"
{kind=link}
Then the OVERLAP software was used in order to suppress those peaks overlaping too much. Four data files are prepared for use by SHELXS-97. The OVERLAP program eliminated reflections having a neighbouring one at less than :
- 0.005° (2-theta) 699 hkl : M33005.hkl
- 0.010° (2-theta) 408 hkl : M3301.hkl
- 0.015° (2-theta) 287 hkl : M33015.hkl
- 0.020° (2-theta) 201 hkl : M3302.hkl
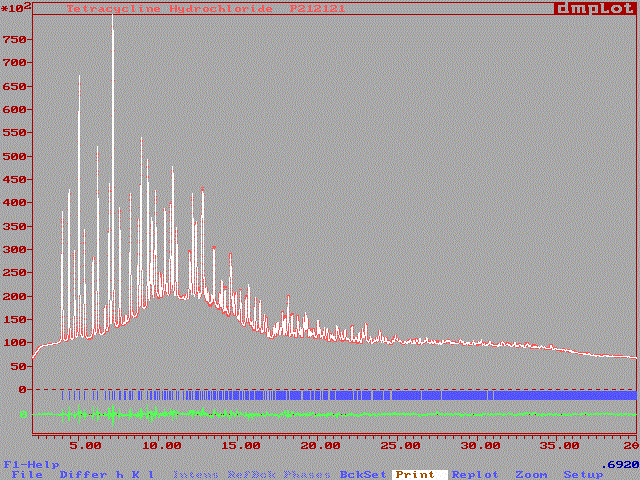
We were expecting 33 non-hydrogen atoms in this structure so that some propositions among the 43 were also discarded on distance criteria : peaks N°21, 26 and 34. Data were used in a SHELXL-97 refinement, the number of atom was progressively reduced to 30 with a R1 reliability factor ~35% (result in tetra.lst). Then a first Rietveld fit was attempted which gave Rp = 4.9%, Rwp = 8.7% (background included) ; Rp=32.6%, Rwp=36.0% (background subtracted) and RB = 28.5%, RF= 17.6%, just refining the scale factor and zeropoint, keeping other profile parameters at their values obtained previously for the Le Bail fit (summary of FULLPROF results in tetra.sum). Then, the atomic coordinates were also refined (C/N/O undifferenciated with C diffusion factor) and the new reliabilities were Rp = 2.28%, Rwp=3.14% (background included), Rp=15.3%, Rwp = 13.2% (background subtracted) and RB=10.08 and RF=13.82 (summary of FULLPROF results in tetra2.sum). At this stage, identification of atoms was attempted, but a mixture of atoms had been obtained from which even a carbon cycle was not located ! As well as the conventional X-ray pattern, the synchrotron one was not enough resolved... Nevertheless, it was thought that methods using special treatment and non-equipartition of the intensity of overlapping reflections could possibly succeed. At least methods of molecule location should succeed. In order to verify this assumption, the PATSEE software was used with the TERMYC coordinates. The molecule was located from both 0.005 and 0.01° OVERLAP reduced dataset. Of course, the rotation and translation corresponding to the best propositions were very small : The refinement of the structure from the coordinates of the isotypical compound (TERMYC in CSD) with synchrotron data is also available here (and see the plot) Rp =9.11 , Rwp = 7.40 % (background subtracted), RB = 4.97 and RF = 8.13%. It was also not possible to refine the tetracycline hydrochloride structure without soft constraints from the synchrotron data (interatomic distances stabilized to unusual values). Of course, no attempt was made for locating hydrogen atoms.
{kind=link}
Finally, a very small single crystal was selected (40x30x20 microns) and was studied at Daresbury. The structure was determined up to the H atoms location and refined without constraint (W. Clegg, to be published). The .cif file will be sent exclusively to the participants.
Thanks for your participation.
Copyright © 1998 L.M.D Cranswick & A. Le Bail