chem cryst
CRYSTALS Software
University of Oxford
-Crystals
+ Research
Contact
Lectures
+ Links
+ Gallery
CSD installation
Chemistry Dept
Division
University of Oxford
chem cryst news
This web page shows some of the new features of Crystals for Windows.
I am extremely grateful to the CRYSTALS users who have
tested and passed on their comments and ideas when working
with the new interface.
I am particularly grateful to Lachlan Cranswick for constantly nagging for
changes, and Ibrahim Tahir and Thierry Maris for being guinea-pigs for
very early versions of the program.
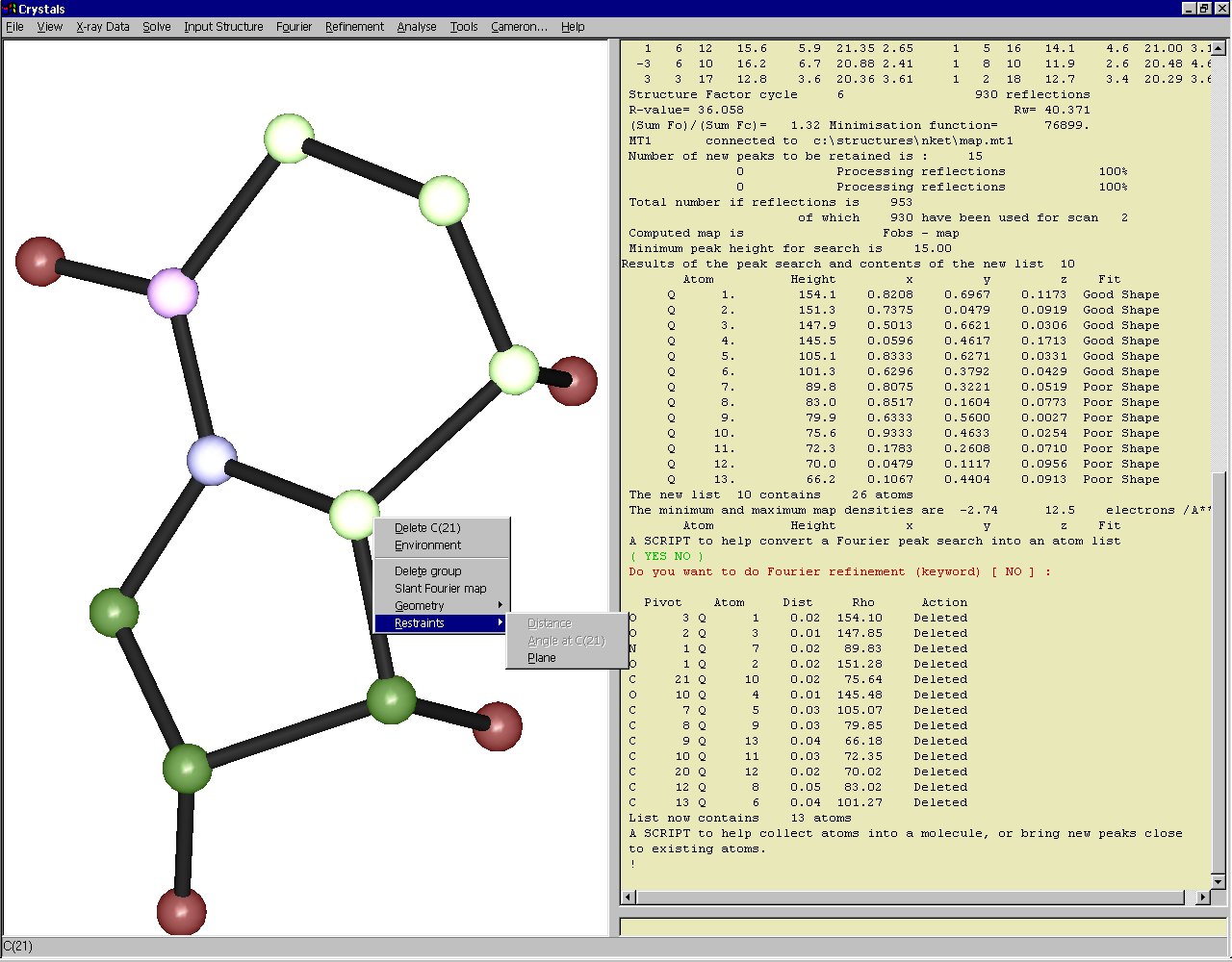
Users can see a representation of their current model structure
at all times. This structures can be rotated and zoomed, and
can also be used to interface with some of the functions of
CRYSTALS.
Nearly all interaction with the user interface is controlled from the
CRYSTALS scripting language, thus making options such as menus
and window layout modifiable and extensible - without having
to recompile the source.
This image shows a context-sensitive menu (right-click on an
atom). Because more than three atoms have been selected (left-
click on an atom) we have the option to restrain the atoms
to lie on a plane.
 All of the old crystals commands are available through the
command line in the main window. An updated version of Cameron
was required to bring it onto the Windows platform. This new
version behaves identically to the old DOS version, but has the
advantage that it can be controlled by the CRYSTALS scripting
language due to closer integration of the two programs.
All of the old crystals commands are available through the
command line in the main window. An updated version of Cameron
was required to bring it onto the Windows platform. This new
version behaves identically to the old DOS version, but has the
advantage that it can be controlled by the CRYSTALS scripting
language due to closer integration of the two programs.
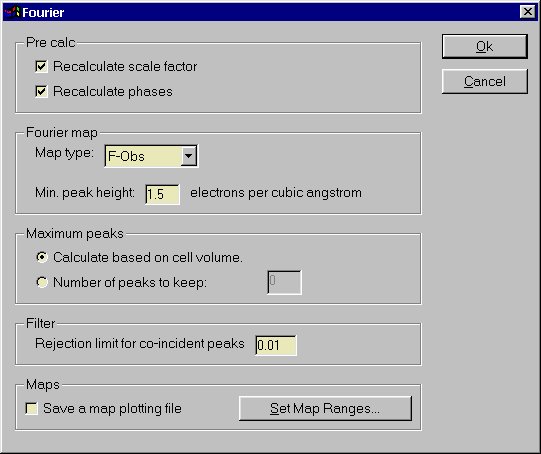 Scripted dialog boxes provide a much more intuitive way to enter
information than the old SCRIPTS 'Question and Answer' method.
The layout, default values and behaviour of all dialog boxes
is controlled by scripts so that they may be adjusted to suit
local languages and preferences.
Scripted dialog boxes provide a much more intuitive way to enter
information than the old SCRIPTS 'Question and Answer' method.
The layout, default values and behaviour of all dialog boxes
is controlled by scripts so that they may be adjusted to suit
local languages and preferences.
 The old ROUTINE script is probably one of the most widely used. For
most structure solution and refinements, it will guide the user to
a publishable structure within a short time. One of the problems with
ROUTINE is that for non-routine structures it is difficult to
fix any problems that occur during an analysis, whilst staying in the
general scheme of this useful script.
The old ROUTINE script is probably one of the most widely used. For
most structure solution and refinements, it will guide the user to
a publishable structure within a short time. One of the problems with
ROUTINE is that for non-routine structures it is difficult to
fix any problems that occur during an analysis, whilst staying in the
general scheme of this useful script.
The new script makes use of the extra options that can be presented
to you using a graphical interface. It uses the same logic as the
old ROUTINE script to decide what to do next, but then offers
this to you as a default choice from a list of options. This gives you
much more freedom to investigate and resolve problems as they occur
in your analysis.
[Download | Copyright | News and Fixes | Bug Report | Mailing list | Documentation | Wiki | Tutorial | Workshops | Screenshots | Obsolete ]

