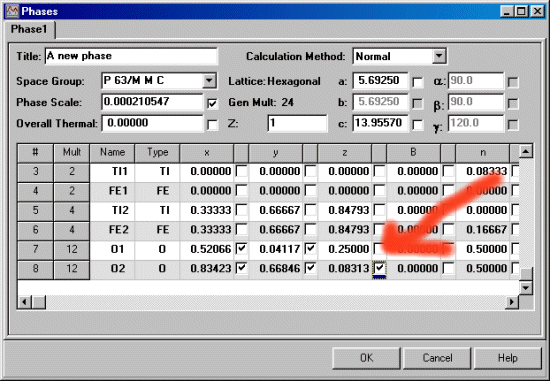
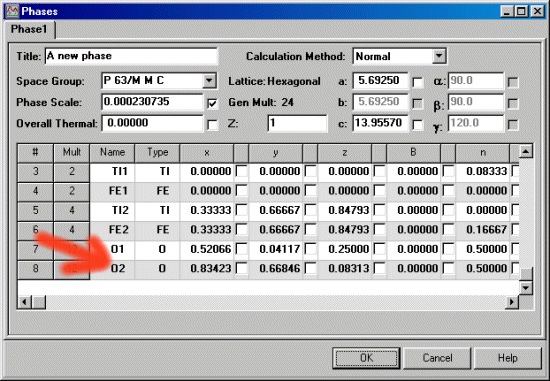
In the following screen dump, the O1 and O2 atoms are both x, 2x, z positions. To correctly refine the Oxygen atom positions, y must shift double to that of x to satisfy the constraints of symmetry for this particular Titanate structure.
|
LHPM-Rietica Rietveld (like DBWS and Fullprof Rietvelds) does not
automatically handle symmetry constraints. Instead the user must
manually implement the symmetry constraints. The following
gives an example of how to do this with x, 2x, z positions.
Click here to download the ZIPPED example data file and input file.
|
|
In the following screen dump, the O1 and O2 atoms are both x, 2x, z positions. To correctly refine the Oxygen atom positions, y must shift double to that of x to satisfy the constraints of symmetry for this particular Titanate structure.
|
|
To set the atom shift constraints via the GUI (Graphical User Interface), first the atom positions must have the refinement flag set (via the Model, Phases menu). Thus the following screen image shows the atom position flags being set to refine for the Oxygens. Note that the 0.25 z position for O1 is not set to refine as this is a special position. You would normally check in the IUCr International Tables as to find out the symmetry constraints of each position.
|
|
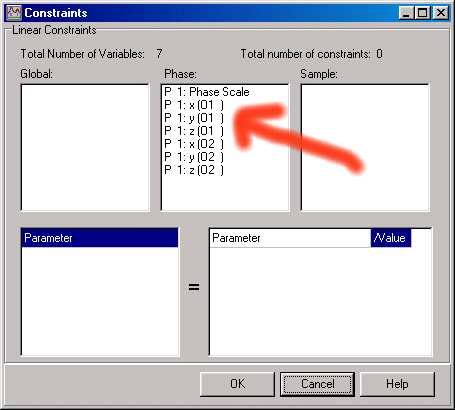
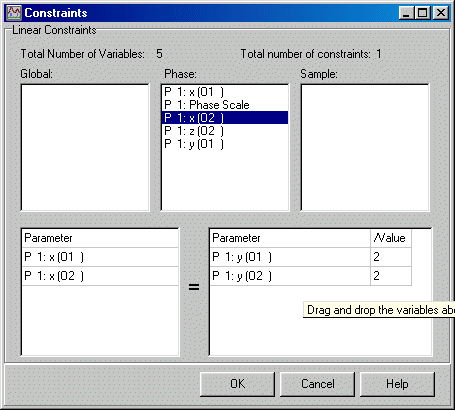
To set x, 2x atom constraints, now enter Model, Constraints. Click and drag via the Mouse is now used to set the constraints from the list of refined parameters.
|
|
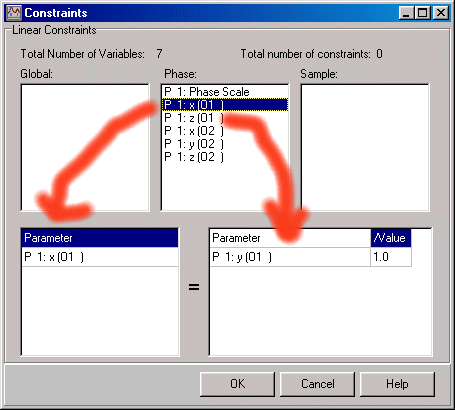
As an example for O1 (Oxygen 1).
However, we are not finished as this sets a linear 1:1 (x, x, z) constraint on the two parameters whereas we need a 1:2 constraint (x, 2x, z).
|
|
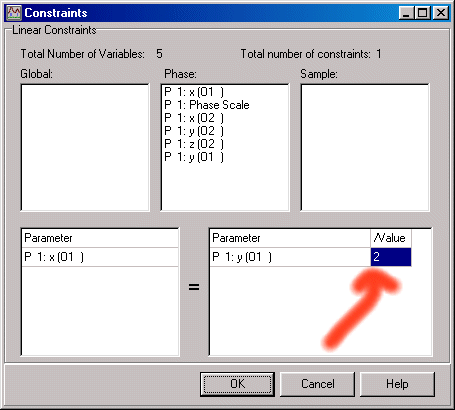
Now, in the bottom right /value box. Set this to 2. This has now set the x, 2x, z constraint.
|
|
Now do the same for the O2 position.
|
|
If you manually peruse the INP input file; you will see the 11/12 code words as the 21/22 codewords that show how these constraints are represented in the input file. (near the bottom of the input file in bold)
|
|
Now that these atom positions are appropriately constrained, you can refine them.
|