|
This example assumes you have done the determination of
the space group using WinGX in a previous tutorial page..
In this tutorial/run-through, only a very minor portion of the functionality of Dirdif using the automatic structure solving PATTY mode. Dirdif, amongst other things, has excellent options for expanding structures and solving on fragments using Patterson search. |
The starting information that is normally known before starting
WinGX is:
|
|
At this point we have already defined the space group and
Now we wish to try and solve this inorganic phase using Dirdif in automatic Patterson mode.
If not already open, run WinGX to bring up the following Menu Bar
|
|
Click on Solve, Dirdif, PATTY menu option to start to try and
automatically solve the structure using Dirdif automatic Patterson methods.
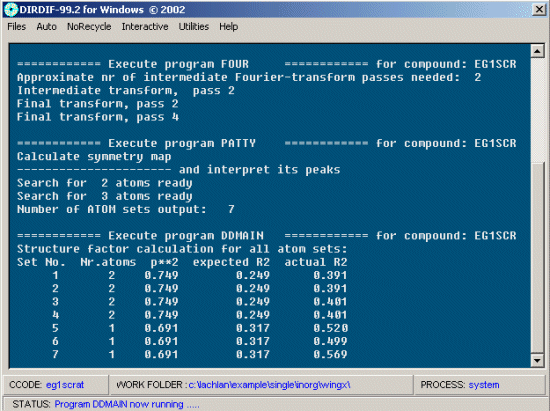
Dirdif then run and try and solve the structure.
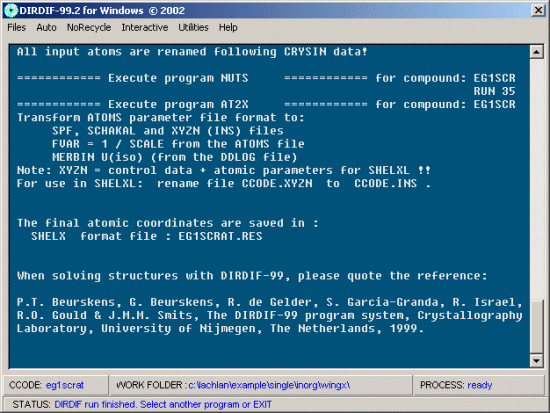
When dirdif finishes, use File, Exit to exit the program in a civilised fashion. Dirdif has also output a Shelx *.res file.
|
|
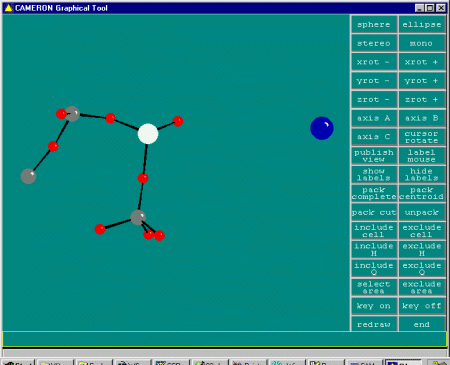
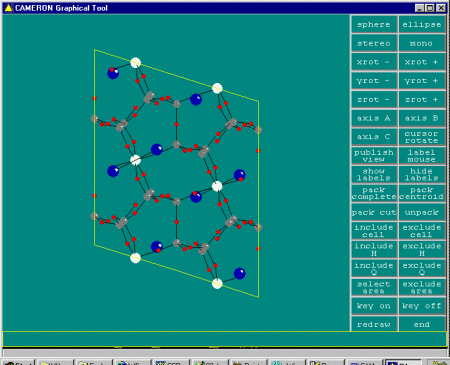
With Dirdif having now run, you can then enter Cameron or another
graphics program of your choice; or examine the output file. In this
case, it suggests the following trial structure solution for you
to check out.
TITL XYZN file = SHELXL INS file, from DIRDIF output for EG1SCR CELL 0.71073 13.38600 7.42300 15.13400 90.0000 107.7100 90.0000 ZERR 4.000 0.00200 0.00200 0.00100 0.0010 0.0500 0.0500 LATT +7 SYMM -X , Y , 1/2-Z SFAC CS O SI TI UNIT 8 60 24 4 L.S. 3 REM use BOND for distances and angles: BOND REM FMAP 3 = electr.dens., FMAP 2: Fo-Fc Fourier FMAP 3 REM Plan n: print n additional Fourier peaks REM Plan -n: print includes connectivity PLAN -10 REM TEMP nn = Temperature of data collect. in Celcius REM TEMP 20 REM SIZE = crystal size in mm : REM SIZE 0.5 0.5 0.5 REM crystal color and shape ? REM Write atoms file in PDB format (with H) : WPDB -1 REM Warning: check HKLF below !! REM ---------------------------- REM 3=Fobs, 4=FobsSQ, > HKL file FVAR 0.33670 CS1 1 0.11670 0.24067 0.40880 11.00000 0.02140 TI2 4 0.25000 0.25000 0.00000 11.00000 0.02140 SI3 3 -0.00071 0.23252 -0.14460 11.00000 0.02140 SI4 3 0.17671 -0.06959 -0.33149 11.00000 0.02140 SI5 3 0.34454 0.03862 -0.14244 11.00000 0.02140 O6 2 0.25164 -0.03664 -0.23045 11.00000 0.02140 O7 2 0.10899 0.19671 -0.07455 11.00000 0.02140 O8 2 0.38048 0.23630 -0.17128 11.00000 0.02140 O9 2 0.29954 0.05392 -0.06000 11.00000 0.02140 O10 2 0.23714 0.07285 0.09110 11.00000 0.02140 O11 2 -0.04929 0.41947 -0.12644 11.00000 0.02140 O12 2 0.00000 0.26946 -0.25000 11.00000 0.02140 O13 2 -0.08424 0.06798 -0.14176 11.00000 0.02140 HKLF 4 END
|