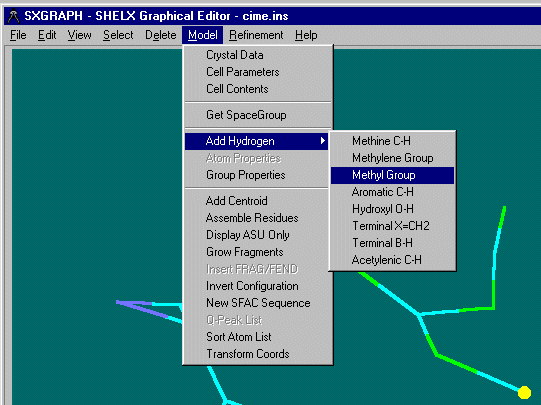
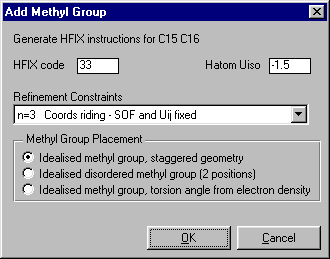
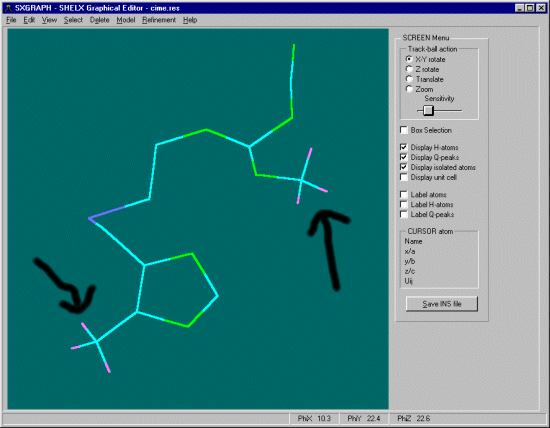
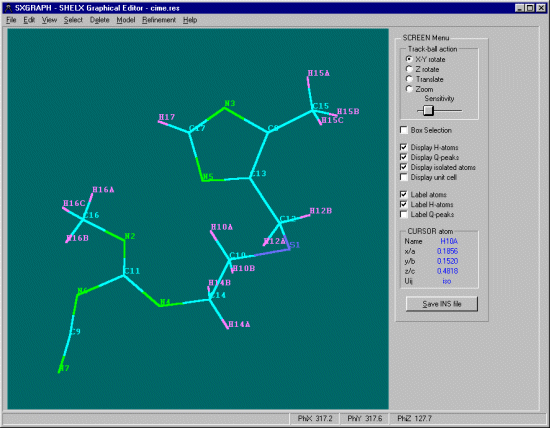
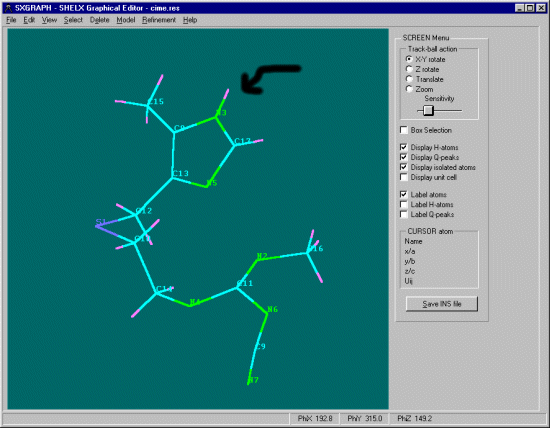
SXGRAPH will allow you to graphically select atoms of interest, then apply the Hydrogen
addition commands. Shelxl97 is then run with zero (0) cycles to generate the hydrogens.
Refer: Perils of automatic hydrogen placement on powder refined structures
A note from David Watkin on this (in the context of Rietveld refinement
before placing the hydrogens):
Date: Mon, 11 Sep 2000 13:46:27 +0100 (BST)
From: David Watkin [david.watkin@chemistry.oxford.ac.uk]
To: Lachlan Cranswick [L.M.D.Cranswick@dl.ac.uk]
I've looked at the cimetidine structure. Instead of trying to put H atoms
on just now, you would be better off using restraints to get the bond
lengths correct. The C4-C17 bond on 1.7 A cannot be right - ie it is
wrong, so should be put right. The C-Cmeth is short, so CRYSTALS thinks it
is SP2, as is the N-Cmeth. A student last year wrote code to put H on N,
which will go into some future release, but guessing N hybridisatioon from
bond lengths is even more risky than C.
More information on "To restrain - or not to restrain"
Date: Wed, 13 Sep 2000 11:18:59 +0100 (BST)
From: David Watkin [david.watkin@chemistry.oxford.ac.uk]
To: Lachlan Cranswick [L.M.D.Cranswick@dl.ac.uk]
This is a deep philisophical problem, and draws in the
Bayesian/non-Bayesian people. The non-Bayesians say that each experinent
should be treated as separate from any other, so you just refine as best
you can with the x-ray data you have. The Baysians say that this is
daft. If some other good experiment has given good values for something,
you should include this knowlwdge in the current process. The trick is to
know what weight to give this external information. A weight of zero makes
it a non-Baysian calculation. A weight of infinity makes it a constrained
calculation. Other weights lead to restraints.
Im a Bayesian myself. If a bond length is weird, either its a fantastic
discovery, or its wrong. If it's wrong - what's the point in publishing
it? Put it right. If the Rfactor goes up, then the data is rubbish and
needs improving. If it stays about the same, then the data doesn't define the
parameter - but it may also fall showing that the trial model was poor. If
one parameter is left at a poor value, others may take on incorrect values
to try to compensate - this is because the parameters are always correllated!
Best wishes
David
In the case of the Methyl Carbons, select these carbons by clicking on them with the mouse
(selected atoms have a yellow dot on them).